This can affect the energy by +- 0.01 kcal/mol
The worst case in the validation suite is the molecule labeled 'JANDOR' which has 53 atoms and 1111 non 1-4 VdW pairs.
There is an inconsistency in the Optimol implementation when it chooses the torsional bond type of certain C-N bonds. To emulate this behavior we need to add the keyword 'MMFF_CN_TORSION_FIX' to 5 molecules: CYGUAN01, DIVJUN, FOYMAH, FULRAF and VEWZOM.
For example in FOYMAH:
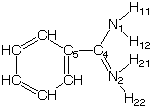
Clearly both Nitrogens are the same, due to delocalization of electrons. Yet, torsions of the form C-C-N-H are different in Optimol's implementation. We believe they should be the same. They are identical for bond-stretch and bend rules. In Optimol two (of the four) have a torsion type of '2', the others are '0'.
Comparing the output describing these torsions for the two
programs (Optimol and Spartan) we see:
--- Optimol (edited from the Validation suite) ---
C5 C4 N1 H11 37-57-55-36 2 .179.931 0.000 0.000 4.800 0.000
C5 C4 N1 H21 37-57-55-36 2 ..-2.064 0.006 0.000 4.800 0.000
C5 C4 N2 H12 37-57-55-36 0 -179.840 0.000 0.000 10.000 0.000
C5 C4 N2 H22 37-57-55-36 0 ..-1.442 0.006 0.000 10.000 0.000
--- Spartan default (edited) ---
2: H21 - N1 - C4 - C5 ==> [ 0.000 4.800 0.000 ] [2] 3
4: H11 - N1 - C4 - C5 ==> [ 0.000 4.800 0.000 ] [2] 3
6: H22 - N2 - C4 - C5 ==> [ 0.000 4.800 0.000 ] [2] 3
8: H12 - N2 - C4 - C5 ==> [ 0.000 4.800 0.000 ] [2] 3
--- Spartan with the MMFF_CN_TORSION_FIX keyword (edited)
2: H21 - N1 - C4 - C5 ==> [ 0.000 4.800 0.000 ] [2] 3
4: H11 - N1 - C4 - C5 ==> [ 0.000 4.800 0.000 ] [2] 3
6: H22 - N2 - C4 - C5 ==> [ 0.000 10.000 0.000 ] 3
8: H12 - N2 - C4 - C5 ==> [ 0.000 10.000 0.000 ] 3
Another example is 'CYGUAN01'
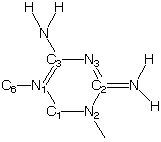
For the torsion C6-N1=C3-N3 and C1-N1=C3-N3 a torsion of type '2' is found. After 2 "rule-fall-backs" Spartan returns a force-constant of V2=4.8, which is the same that both programs get for C6-N1-C3-NH2. However, Optimol chooses a torsion type of '0' and V2=10.0.
Note that this difference is usually undetectable as the NH2 group is nearly planar and differences only show up in strained systems.
If the 'MMFF_CN_TORSION_FIX' keyword is not applied, the energy differences between the 'mmd' structures and the unminimized Spartan energies for the 5 molecules in question are:
Molecule |
Published Energy |
Energy Without "Torsion Fix" |
Delta |
Energy With "Torsion Fix" |
|---|---|---|---|---|
| VEWZOM | -9.37968 | -9.379853 | .00017 | -9.379675 |
| FULRAF | 98.97744 | 98.976814 | .00062 | 98.977440 |
| FOYMAH | -4.87726 | -4.880598 | .00333 | -4.877264 |
| DIVJUN | 86.58284 | 86.577870 | .00497 | 86.582836 |
| CYGUAN01 | -254.74397 | -254.773534 | .02956 | -254.74396 |
Spartan produces identical results for the dative and hypervalent test suites.
We, (Wavefunction), have a number of customers who also use 'delocalized' bonds to represent delocalized charge and aromaticity. (i.e. Sybyl's type '5' bond, drawn with 2 lines, one solid and the other dashed.)
Our goal is to get these representations to match correctly. This is
problematic, especially in the case of fused rings as it confuses
the 'upgrading-to-aromaticity' rules. For example; making sure the
central ring in DAKCEX remains non-aromatic is difficult:
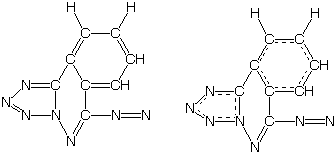
Thus, we advise customers use the Keukule
drawings of aromatic systems, unless one wants to specifically
override the MMFF94 aromaticity rules.
Other, non-aromatic delocalized charge distribution,
(ie. SO2, NO2 and NCN+) should be ok.