Quantum Mechanics Energy FAQ
- How do I calculate the heat of reaction?
Quantum chemical methods can be used to calculate heats of
reactions. The Heat of formation (HOF) is a special type of
reaction.
For example, to calculate the bond separation energy of
CH3CH2NH2 + CH4 -> CH3CH3 + CH3NH2
one would perform
4 energy calculations (one for each molecule) and subtract
the energies.
E-reaction = E[CH3CH3] + E[CH3NH2] -
(E[CH3CH2NH2] + E[CH4])
The dialogue (Display menu > Reactions)
is a great tool for
easily calculating this type of equation,
see Stereospecific Diels-Alder Reactions tutorial
(Activities menu > Tutorials > Chemical Reactions chapter)
for an example.
Experience has shown that the quality of the calculated
heats of reaction depends on both the method used to calculate
the energy (E[..]) and the type of reaction.
Our guide book explains that the
"best" type of reaction is an isodesmic reaction. (An
isodesmic reaction
is one in which the number and type of bonds remain the same
during the reaction. For an example see the discussion
on basicities.) The book also lists other types of
reactions and gives quantitative examples of the accuracy one
can expect for different theories and basis sets.
Return to Top
- How do I calculate the heat of formation?
In principle the heat of formation (HOF) is calculated like
any other reaction, see the discussion above.
Sadly, the experimental HOF consists of some of the most
difficult reactions to calculate: breaking all bonds and creating
atomically pure compounds such as H2 for hydrogen and
graphite for carbon.
2(CH3CH2NH2) -> 7(H2) + N2 + 4(C-graphite)
Thus, good HOF calculations usually
require methods better than QCISD with very large basis sets.
These can be extremely time-consuming, even for molecules of only
a few heavy atoms!
The G3 method is a good place to start if you
are interested in accurate Heats of Formation.
Chances are that if you think you want the HOF formation,
you may only need to compare the relative differences in energy
of certain reactions, and can use a less time-consuming approach than
the methods required for Heat of formation calculations. The
guide book
includes many examples of HF/6-31G* without vibrational
corrections performing well (against experiment). The discussion on
finding the *best* energy also covers
this and other common theory levels.
Return to Top
- What is the G3 method?
The G3 method (the most used of the Gx series)
is a time consuming recipe for
calculating accurate "Heats of Formation" (HOF) from first principles.
G3 consists of 10 calculations:
- HF/6-31G* optimization
- HF/6-31G* frequency and vibrational free energy
- MP2(full)/6-31G* geometry calculation
- MP2 and MP4 6-31G* single points
- MP2 and MP4 6-31+G(d) single points
- MP2 and MP4 6-31G(2df,p) single points
- QCISD(T) 6-31G(d) single points
- MP2(full)/6-311+(2df almost) single point (the G3large basis set)
- A spin-orbit correction term.
- An empirical "higher level correction" for valence electrons
All these energies are combined, along with similar
calculations done for each atomic species. The result is
then subtracted by experimental energies of the "standard
state atomically pure" systems to arrive at a good
approximation for the actual HOF. A quick overview of how these
steps are used can be found in our
discussion of calculating the exact
energy.
Note that each of these steps is relatively time consuming
and practical only for very small systems.
The G3(MP2) method is often more than twice as fast, but
still prohibitively time consuming for all but small molecules.
The reference to the G3 papers can be found
at the end of this FAQ.
Return to Top
- What is the T1 method?
The T1 method is a simpler recipe to calculate the absolute
heat of formation. It is similar in concept to the
G3 method but replaces the post MP2
calculations with empirical relations based on Mulliken bond
order and implements a faster MP2 approximation. Thus it is much
faster than G3 or G3(MP2) and can be applied to much larger
molecules. The steps of the calculation are:
- HF/6-31G* optimization
- RI-MP2/6-311+G(2d,p)[6-311G*] single point energy. This is
a dual basis set approximation to the MP2 energy
- An empirical correction using the HF/6-31G* Mulliken bond
orders to capture the majority of post MP2 energy, and
bond vibrational energy.
This is discussed in full in the defining paper.
Return to Top
- What is the "Enthalpy of Vibration"?
The quantum mechanical calculations in Spartan assume that
molecules are isolated, with a temperature of zero Kelvin, and
with stationary nuclei. Real experiments are carried out with
vibrating molecules at finite temperature (often 298.15 K). In
order to correct for these differences Spartan can use
calculated
frequency data to determine a set of normal-mode vibrational
frequencies (vi). (These are the same
data used to generate IR spectra.)
Using this approximation, energy corrections are
fairly straightforward. Splitting the Enthalpy correction into
four (4) parts to include the 'Zero point energy' (Zp),
the temperature correction (Hv), enthalpy due to
translation (Ht) , and enthalpy due to rotation
(Hr).
Zp = (1/2)*sum[ vi ]
Hv = N*h*sum[ vi / (
eh*vi/kT - 1 ) ]
Ht = (3/2)RT
Hr = (3/2)RT (or RT for linear molecules)
Note that for small molecules this is a very good
approximation. However as molecules get larger the
'normal-mode' approximation becomes less valid. For example:
- floppy side groups, (such as methyl rotors) behave like a 3 well
system, not the single well assumed in the normal-mode
approximation,
- anharmonic effects become more important,
- large scale flexing motion, which is difficult to accurately
calculate, begins to dominate the energy correction.
- conformational flexibility becomes an issue which (energetically) may be
more important than the vibrational temperature correction (Hv).
These problems are magnified if one is examining the
entropy or
free energies of larger molecules. For these reasons low
frequency vibrations are often systematically ignored.
Return to Top
- How is the entropy calculated in Spartan?
The entropy in Spartan is calculated from the same data as
vibrational enthalpy (Hv). The
equation for entropy (S) can be written as follows:
S = Str + Srot + Svib + Sel
Str = nR{(3/2) + ln[ (pi*MkT)3/2*(nRT/P) ] -
ln(nNo) + 1}
Srot = nR{(3/2) + ln[
(pi*vA*vB*vC)1/2/s ]
Svib = nR*sum[
(ui*eui-1)
-1 - ln(1-eui)]
Sel = nR*ln[ "electronic ground state degeneracy" ]
The constants ui are proportional to the
vibrational frequencies and vA are inversely
proportional to the moment of inertia of the rigid body. It is
important to note that these equations ignore any entropy due to
conformational flexibility, which may dominate for
larger molecules. Also note that low frequency vibrations will
dominate the calculation of entropy. It is these
modes which break the normal mode assumption, thus making
entropy calculations of larger molecules suspect.
Return to Top
- How can I calculate basicity?
You can use the isodesmic proton transfer reaction relative
to a standard to calculate the relative basicities. For example
if we choose the ammonia molecule as a standard we would write:
BH+ + NH3 -> B + NH4+
where B is the compound of interest. Then the
relative proton affinity is:
E[BH+] + E[NH3] - E[B] - E[NH4+]
Relative proton affinity is a measure of the basicity which
can easily be converted to pH.
In our methods book (table 6-17)
we show the accuracy
of this calculation for a number of methods and basis sets
for series of Nitrogen Bases. A regression analysis shows that
even simple HF/6-31G* methods provide results with an accuracy of
less than 4 kcal/mol on a system covering 125 kcal/mol range.
There is also an interesting example of predicting pKa's from
the graphical map of the electrostatic potential. This can be
found on page 478 of A Guide to Molecular
Mechanics and Quantum Chemical Calculations.
Return to Top
- How can I calculate pH?
The pH of a compound should be related the
energy (enthalpy) of the reaction described
above
BH+ + X -> B + XH+
If you know the pH of a few standards you can use
these in a regression fit (using regression tools in
Spartan's spreadsheet).
This will result in a simple linear relation
correlating 'energy' with 'pH'.
Return to Top
- Advice on calculating pKa's
While pKa's can be calculated using methods described above
for pH and basicity
it is often possible for to use empirical relationships.
For example the Groups of Organic Molecules tutorial (menu > Tutorials) has an example of determining the
pKa of carboxylic acids
from the electrostatic potential of the acidic hydrogen. With a
simple linear regression one can get rather good prediction for a
rather limited case of molecules. As is shown in the first plot below
which is the result of going through the above mentioned tutorial.
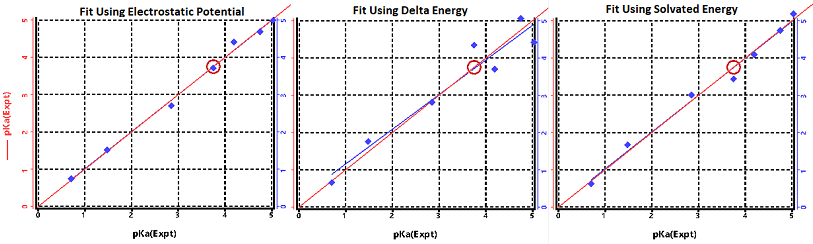
For many cases empirical rules such as this work "good enough"
and are so quick to calculate that this is the recommended approach.
In principle we calculate the pKa from first principles
using the deceptively simple equation
pKa = ΔGaq / [ln(10)*RT] .
Unfortunately one typically requires very high theory levels.
For example Thapa and Schlegel show that for thiols not only
is an expensive method and basis set required, but a solvation model
including implicit and explicit water molecules is needed.
(The Journal of Physical Chemistry A 2016 120 (28), 5726-5735)
However, using the fact that the change in energy should be
proportional to the energy of de-protonation one can do some quick
calculations to approximate pKa's. The second plot in
figure above is a linear fit of the same carboxylic acids but
calculate the energy difference between the neutral acid, and
the de-protonated acid.
In this example the geometry used was the same as the neutral case,
except the acid hydrogen was removed.
In some cases this kind of fit might be "good enough".
The third fit is a similar calculation but done in solvent.
Return to Top
- Spartan generates many different energies. I want the best one.
Which one is the best energy?
Science is a series of approximations. The trick is to use
the largest simplification one can and still get reasonably
'good' predictions. This said, we always feel more comfortable
with a theory if a recipe exists to get the *exact* answer.
In QM calculations this is so prohibitively time consuming as to be
impossible, but it can be described. We describe this 'exact'
calculation in the next section.
One definition of the *best* energy is the energy which answers
one's questions with enough accuracy, yet consumes the minimum amount of
time and resources. As mentioned in the discussion on
calculating heats of reactions, this is often
achieved by writing a near-isodesmic reaction and calculating an
energy difference. (The key here is the 'energy difference'.
It allows cancellation of systematic errors.)
In order to find what is *best* for your question, we
suggest starting with a question/reaction in which you
know the answer and that is similar to your reaction of
interest.
Try this reaction with HF/6-31G* or HF/3-21G*.
If that isn't accurate enough then add the zero-point energy (ZPE)
corrections.
(scaled by .90 for HF or by .99 for DFT)
If that fails, try MP2 or DFT energies with larger basis sets.
If that fails attempt single point CCSD with ZPE corrections.
Finally try the T1,
G3(MP2), or G3 methods.
Conversely, if HF/6-31G* is good enough, you may want to try
faster methods to see if they are also acceptable. HF/3-21G* is a
popular basis set and much faster than 6-31G*. Semi-Empirical
is the simplest method using quantum mechanics approaches while
requiring an order of magnitude fewer resources and time.
Molecular mechanics methods, while limited to ground states of common
chemistry groups, is extremely quick and in the areas in which
it is parameterized can be fairly accurate.
If you are new to molecular modeling,
Wavefunction's guide book,
discusses many types of reactions and molecular properties,
quantitatively comparing many different theory levels.
Return to Top
- How can I calculate the most precise/exact value
for the energy? Or,
18 steps to calculate the exact free energy of a molecule.
The complete Enthalpy (H) and the Gibb's Free Energy
(G) of a given conformer of a
molecule in the dilute gas phase can be written
H = Ee + Zo + Hv + Ht + Hr + pV
G = Ee + Zo + (Hv+Ht+Hr) - T*(Sv+St+Sr) + pV
where;
- Ee is the complete electronic energy including
- an infinite basis set.
- including all electron correlation (a full CI
calculation)
- accounting for spin-orbit coupling and relativistic
effects.
- Zo is the zero point energy of the nuclei.
- Hv is the enthalpy due to vibrations
(beyond the zero point energy).
- Sv is the entropy due to vibrations.
- Ht, Hr, St and Sr are the
enthalpy and entropy due to rigid body rotation and translation.
Calculating all of these terms is impossible for the foreseeable
future for any real/non-trivial molecule. However a number of
methods and recipes have been proposed that attempt to approach
this limit.
Below we describe such an algorithm using the
G3 method.
The G3 method is a specific
thermochemical recipe
available in Spartan that attempts to produce
accurate results in a relatively short amount of time. By
studying it
we can generalize it to imagine a method which should give the
*exact* results even if such a recipe is impractical.
Below is the edited result of a G3 run on a water molecule,
used as a reference:
Energy HF/6-31G(d) -76.009809 1
Energy MP2/6-31G(d) -76.196848 2
Energy MP4/6-31G(d) -76.207326 3
perturbation correction -0.000565 4
polarization correction -0.074488 5
diffuse correction -0.012979 6
basis set correction -0.081653 7
spin-orbit correction 0.000000 8
electron pair HLC -0.025544 9
---------------------- ------------------
G3 energy (Ee) -76.402556
HF/6-31G(d) zero point 0.020515 11
G3 energy (Eo) -76.382040
HF/6-31G(d) Vibrational 0.020518 12
HF/6-31G(d) Translation 0.001416 13
HF/6-31G(d) Rotational 0.001416
RT constant 0.000944
G3 kT energy (H298) -76.378260
Atomic Energy -76.032990 14
dH(298) -0.091381
dH(298) -57.34 kCal/mol
A description of successive improvements to the energy is provided. The
details of each step are not discussed here, as the main point is
to
provide an overview of what is required for an
*exact* answer.
- First we start with the HF energy using the 6-31G(d)
basis set. This is the total energy required to pull
the 10 electrons, 2 protons, and 1 Oxygen nuclei apart.
HF methods ignore electron correlation energy. The goal of DFT
and other post HF methods is to account for this electron
correlation energy.
- MP2 can often
correct for 80% of this energy. (G3 minimizes
the coordinates at the MP2 level. All further energies
are done with this geometry.)
- MP4 is an improvement to MP2 collecting almost 95% of the
total correlation energy.
- In an attempt to do better than 95% of the correlation
energy (from MP4), another correction, the "perturbation correction", is made. (G3 theory uses a QCISD
calculation for this term).
One weakness in the approach described so far is the basis set.
There are a number of possible improvements that can be made to
the basis set:
- Adding polarization terms is often the easiest way to
improve a basis set.
- Diffuse functions are sometimes useful, especially for
negatively charged systems.
- We also need to make the core basis set more flexible
with additional terms.
- We have so far totally neglected any spin-orbit contributions
to the energy so we may want to add this energy term in.
- There are other systematic electronic errors.
The G3 method
lumps these into a "High Level Correction" (HLC) term.
- We could also add relativistic effects to the energy although
this is not done in the G3 method.
If we carry all the above corrections correctly to their
infinite limit we should have the *exact* electronic energy.
We label this energy Ee.
But this would still not be good enough. We've made the
assumption that the nuclei do not move. Quantum mechanics
tells us that everything is moving, even at absolute zero; the so
called "zero point" energy (Zo). By doing a normal mode
analysis we can calculate this zero point energy.
- G3 calculates this zero point energy at the HF level with
the 6-31G(d) basis set. If we had infinite computational
resources we could improve this in the same way we discussed
improving the electronic energy.
The above energy would be correct at zero Kelvin, but chances
are that the system we are studying exists at a non-zero temperature.
We can make normal mode corrections to the energy, which we
now call enthalpy and label it H298. (298.15 K being the
most common temperature of interest.)
- G3 adds the
enthalpy (Hv) due to
normal mode vibrations.
- G3 also adds energy due to translating and
rotating the entire molecule, as well as a pressure term
from the ideal gas law equation (RT==PV).
At this point we notice that the energy is in some "strange"
units called Hartrees. In order to compare with other results
it is useful to do a conversion.
- G3 does an energy conversion to kcals, attempting
to define zero using the standard states of the constituent atoms.
This finally is dH (delta-H). This is the final goal of
the G3 theory.
This final conversion step is not always necessary.
What is necessary in QM
calculations is to ensure that when comparing energies, the
energies are not only in the same units, but define the same
zero. (Typically that means the same basis set and theory
level, and molecules with the exact same constituents and
number of electrons.)
Often, when examining reactions we require even more; the Gibbs
free energy (G) which is calculated by subtracting
entropy:
G = H - TS + PV.
- A first approximation of the entropy (S) can be
calculated using the same
frequency data as Hv was previously.
-
In our calculation of H298 and S we have used
the normal mode approximation
ignoring anharmonic terms. These actually become important
at higher temperature, and are often most noticeable in the
calculation of the entropy. Anharmonic cross terms may also
become important. For low frequency vibrations the normal
mode approximation may not be appropriate and other equations
may need to be examined to model rigid rotors (such as methyl
groups) or low frequency bending and folding modes.
- For flexible molecules it is necessary to look at multiple
conformers of the same molecule. In a goal to get the
*exact* answer one would need to apply all the previously
discussed steps to each conformer and weight their
contributions using a Boltzmann weighting scheme.
The above steps should cover all the physics of a
completely isolated molecule. (i.e. In a vacuum or a dilute gas.)
Adding a real environment (such as a solvent or a crystal
lattice) is vastly more difficult.
- Calculating this environment could be attempted by adding
a shell of the environment's atoms surrounding the molecule
and performing all of the previous steps on this larger system.
Combining all of these corrections terms "correctly"
(for which there is currently no 'good' systematic procedure)
one should arrive at the "exact" answer.
Thankfully, a careful examination of the types of chemistry questions one typically asks shows that one does not need to do all this
work. (See our guide book for
a thorough discussion of this.)
Surprisingly, in many cases, HF/6-31G(d) is a good
approximation to reality.
This happy accident makes quantum chemistry possible on today's
computers.
Spartan gives users the ability to choose from a number of advanced
correlated methods, and implements the G3 recipe, which is a
standard approach for improving the accuracy of calculated energies.
Spartan also provides the flexibility to work with a wide variety of
theoretical models:.
- Steps 2-4: A number of post HF methods are
available, including
- DFT: BP, EDF2, B3LYP, M06, wB97X-D, wB97X-V (and many more)
- MP2, RI-MP2, , and MP3
- CCD, VOD, VQCCD, QCISD, CCSD, OD, QCCD
- MP4SDQ, MP4
- QCISD(T), CCSD(T), OD(T)
- VQCCD(2), VOD(2), QCCD(2), CCSD(2), CCD(2), OD(2)
- Steps 5-7 Spartan has a full range of basis sets
available, and allows the input of external basis sets. These
include:
- STO-3G,3-21G(*)
- 6-31G with flexible polarization and diffuse options.
- 6-311G with flexible polarization and diffuse options.
- cc-pVDZ, cc-pVTZ, cc-pVQZ, cc-pV5Z
- the def2 series of basis sets
- the G3 basis sets; G3MP2large, G3large
- ECP basis sets including LACVP and LANL2DZ
- and the ability to add user basis sets.
- Steps 11-15 The ability to calculate frequencies and
other thermodynamic data allows for the calculation of free
energy due to vibrations using the normal mode approximation.
- Step 17 Spartan's conformation module makes it
easy to generate and work with multiple conformers.
- Step 18 Spartan has a number of
solvation methods
which can be used to approximate a solvent.
Return to Top
Why do Molecular Mechanics, Semi-Eempirical, and Ab initio
energies look so different?
All three approaches have a different definition of zero, and
use different units (by convention).
- Molecular Mechanics calculates a "strain" energy. The zero of
this system is one in which all bond angles and lengths are at
their 'ideal' values. This 'zero' may not be
attainable. For example, the water molecule has a 'zero' energy
at the global minimum, however, methane does not (in MMFF).
This is because in methane the 'unstrained' angle is not 104.9
even though the 3-dimensional interactions of the 6 angles
cause the minimized structure to have bond angles of 104.9.
Given this definition one cannot compare molecular mechanics
energies of different molecules. This restriction holds
true even for
different isomers. One can compare energies of
different conformations. In fact this is one of the most
popular and appropriate uses of molecular mechanics.
Semi-Empirical methods use the same definition as
ab initio, but a constant is subtracted for each atom in the
molecule. The goal of this 'shift' is to approximate a heat
of formation (HOF). In reality, this approximation is not
very accurate and should only be used as a first
guess at the HOF. Semi-Empirical methods are most useful for
predicting rough geometries and transition states quickly.
Ab initio methods (including HF and DFT) compare the
energy of the combined molecule with the energy required to
remove every electron, and separate every nuclei. While this
reaction is not a very realistic experiment, it is
theoretically simple and can be used to calculate energy
differences of conformers, isomers, and entirely
different molecules. See the discussion on
calculating the heat of formation
for a more detailed description of using this energy.
By convention ab initio results are returned in units of Hartrees.
See the
units section to
convert from Hartrees to more common units
Return to Top
How many parameters are there in each method?
Each method is different, but an overly short answer would be:
- no parameters for ab initio methods (HF, MP2, MP4, CCSD,
...)
- about a dozen parameters for DFT methods (SVWN, B3LYP,
wB97X-D, ...)
- dozens for Semi-Empirical methods (AM1, PM3, ...)
- hundreds for molecular mechanics methods (SYBYL, MMFF,
...)
For an exact number one will need to examine the primary
literature on these methods. However, slightly more detailed
discussion of these methods presented below. More details can
be found in Spartan's documentation.
- The HF, MP2, MP4, CISD, CISD(T) can be thought of as keeping
more terms in a "Taylor-like" series expansion of the
full/exact quantum mechanical calculation. They are called
'ab initio' because there are no parameters other than
where to/how to stop the expansion.
- The basis sets (3-21G*, 6-31G* ...) can be thought of as a
"Taylor-like" series expansions of the exact electron density.
The smallest have ~10 terms per atom ... and more are added as
the sets get bigger.
The authors of these basis sets would not call them free
parameters, but defend that most of these parameters are
derived and claim there are only a few free parameters that
describe how the expansion is terminated. (In QM parlance;
splitting and polarization.)
- DFT methods can be considered an extension to HF methods,
with a few parameters added to attempt to go from HF to the
exact quantum mechanical solution. In reality these
approximations appear to improve the base HF to MP2 level or
better.
- Semi-Empirical, is a radically abbreviated "Taylor-like"
expansion of the exact quantum mechanical solution. To
compensate for how bad this early termination is, about 20
parameters per atom are added.
- Mechanical force fields are purely empirical; roughly 5
parameters per "atom type", many more for each bond,
(including torsional, bond bending, cross terms). In modern
force fields there are more "atom types" than one might
initially guess. For example, MMFF94 has 8 different kinds of
oxygen. Using the PRINTLEV=4 keyword will dump the parameters
for a given molecule. (Only available for the molecular mechanics
module).
Return to Top
Can I add other DFT functionals?
Spartan can access a large number of DFT functionals.
Only the most popular and well tested are available from the
"Density Functional" pull-down menu of the Calculations dialogue.
For example Truhlar group's M06-L function while not
available by default from the
menu can be accessed by clicking on "More..." at the
bottom of the menu list. This leads to a hierarchical
dialogue offering a wide range of functionals organized
by classes. Additionally, specific functionals may be typed
in the "Options" line of the Calculations dialogue.
When the Enter/Return key is struck the specified functional
will disappear from the Options line and will appear as
the selected functional in the "Density Functional" pull-down menu.
It is possible to define your own functionals with the
EXCHANGE= and CORRELATION= keywords.
Each functional must be followed a colon (':') and then its
weighting. Different functionals are then separated by
commas. For example a functional consisting of 9 parts
"Slater" exchange and 1 part "Becke" exchange would look
like:
EXCHANGE=SLATER:0.9,BECKE:0.1
If any correlation functionals are required one would use
the CORRELATION keyword. For example:
EXCHANGE=HF:0.5,SLATER:0.08,BECKE:0.72
CORRELATION=VWN:0.19,LYP:0.81
The values allowed exchange functionals are
- HF Fock exchange
- Slater Slater (Dirac 1930)
- Becke Becke (1988)
- GILL96 Gill (1996)
- GG99 Gilbert and Gill (1999)
- B(EDF1) Becke (uses EDF1 parameters)
- PW91 Perdew
- PBE Perdew-Burke-Ernzerhof (1996)
- BR89
- B97 Becke97 XC hybrid (1997)
- B97-1 Becke97 re-optimized by Hamprecht et. al. (1998)
- B97-2 Becke97-1 optimized by Wilson et. al. (2001)
- B3PW91
- B3LYP the popular hybrid of Vosko, Wilk and Nusair
- B3LYP5 #5 hybrid of Vosko, Wilk, and Nusair
(Gaussian's version of B3LYP)
- EDF1 also EDF2
- BMK BMK hybrid
- M05 also M052X,
- M06 also
M06HF, M062X,
The correlation functionals available are
- none (no correlation)
- VWN Vasko-Wilk-Nusair #5
- VWN1RPA
- LYP Lee-Yang-Parr
- PW91 GGA91, Perdew
- PW92 LSDA 92 (Perdew and Wang)
- PK09 LSDA (Proynov-Kong)
- PK06
- LYP(EDF1) from the EDF1 functional
- BNLC
- B94 Becke (1994)
- B95 Becke (1994)
- P86 Perdew (1986)
- PZ81 Perdew-Zungner (1981)
- PBE Perdew-Burke-Ernzerhof (1996)
- WIGNER Wigner
- wB97,wB97X,wB97X-D the correlation part of the
B97 series
- LP Liu-Parr (1996)
- HF HF exchange (K), (required for hybrid density
functionals)
- (B88)OP OP correlation optimized for B88 exchange
- (PBE)OP OP correlation optimized for PBE exchange
-
- Wigner
Spartan'18 (utilizing the Q-Chem 5.X code) also includes:
- Exchange: optB88, PBE-GX
- Local XC: HLE16, KT1, KT2, KT3
- Hybrid XC: CAM-QTP00, CAM-QTP01, r CAM-B3LYP, WP04, WC04, TPSS0
- Double hybrid XC: DSD-PBEPBE-D3, DSD-PBEP86-D3, DSD-PBEB95-D3,
PTPSS-D3, PWPB95-D3, B2GPPLYP, PBE-QIDS, and LS1DH-PBE
For those who are familiar with the
Q-Chem Exchange/Correlation notation;
using EXCHANGE or CORRELATION without any
colons sets the appropriate "REM" values.
With colons included, an
"XC_FUNCTIONAL" section is created.
Return to Top
The B3LYP coefficients look different;
are they correct?
We implement the main equation of the Vosko, Wilk, and Nusair
paper. Sometimes, people will use equation #5 of this paper,
which, in Spartan is called B3LYP5.
In some references the coefficients are written differently.
For example we write the B3LYP functional as
EB3LYP =
0.20*ExHF +
0.08*ExSlater +
0.72*ExBecke +
0.19*EcVWN +
0.81*EcLYP
Some literature might have it written (equivalently) as
EB3LYP =
(ExSlater + EcVWN ) +
0.20*(ExHF - ExSlater ) +
0.72*(ExBecke - ExSlater ) +
0.81*(EcLYP - EcVWN )
in order to highlight the "correction to SVWN". Another example is:
EB3LYP =
0.20*ExHF +
0.80*ExSlater +
0.72*(ExBecke - ExSlater ) +
1.00*EcVWN +
0.81*(EcLYP - EcVWN )
Another subtle difference between different B3LYP calculations
can be the selection of the numerical grid used to calculate the
density. See "What size grid does Spartan
use for DFT calculations?" for a discussion of this.
Return to Top
I've heard that "standard" grids aren't good enough. What grid should I use?
Recently there has been some large scale coverage of potential problems
with grid sizes in DFT calculations.
For example a paper by
Bootsma and Wheeler
and mentioned in a C&EN article.
We think our defaults are good for most applications,
but it is "easy" to check by re-running with the HUGEGRID
keyword on sample "energy differences"
(such as the two end-points in a reaction) to confirm.
We determine default grid sizes based on what give consistent gradients,
as opposed to "best energy", but regardless the grid error is nearly always less
than the "incomplete basis set" error, both of which are presumably random
for reaction energies (and other "relative" values, ie. not atomization energies)
Meta functionals like M06* and wB97XM-V do require bigger grids sets
than wB97X-D and other "2nd generation" sets.
(even though we use (70,302) similar to wB97X-D's default to follow Truhlar's paper)
wB97X-D which uses (75,302) by default requires more than B3LYP
for which we use the SG-1/SG-0 grids
(which themselves were optimized for the Pople basis sets 6-31G and 6-311G).
We contend that "grid errors" should be given as "error per atom" dEg/a
(read as delta Energy-grid per atom). Assuming that this
error it is not systematic, the "total" error/noise would then be
[dEg/a] x [N1/2]
The curious might want to examine
the discussion of
the grids we are most familiar
with (SG1 and SG0) which we use for B3LYP and EDF2.
Most modern papers covering "new" functionals will spend time discussing the
preferred grid.
Of course in Spartan you can easily change the default gradi as discussed
below.
Return to Top
What size grid does Spartan use for DFT
calculations?
For the "simpler" functionals, such as B3LYP and EDF2,
Spartan employs a hybrid SG-0 and SG-1
grid as follows: SG-0 for H,N,C, and O,
SG-1 for all other
atoms. For the more "modern" functionals we use a larger
grid (because they are more sensitive to small changes in grid).
Specifically, we use the "BIGGRID" (70,302) for the Truhlar
functionals and Head-Gordon
functionals such as:
Following are some keywords which can be typed into
the Options line of Spartan's Calculations dialogue to change
the grid:
- SG-1 and SG-0, small grids tuned for the
6-31G* basis sets.
- SMALLGRID (Spartan's default for "simple" DFT functionals;
SG-0 for H,C,N, and O and SG-1 for all other atoms)
- EMLGRID (50,194)
- BIGGRID (70,302)
- VERYBIGGRID (100,434)
- FINEGRID (99,590)
- HUGEGRID (250,947)
In the above notation the first number is the number of
shells in the radial direction, the second number (i.e. 194,
434 etc.) is the number of Lebedev radial points.
One can also use the BIGGIRD keyword with an equal sign, to
enter a Q-Chem like grid notation. Specifically a 12 digit
number with the first six (counting leading/implied zeros)
defining the number of shells in the
radial direction and the next 6
defining the number of Lebedev radial points.
i.e., BIGGRID and BIGGRID=70000302 both refer to
(70,302)
Valid values for Lebedev grids are:
6, 18, 26, 38, 50, 74, 86, 110, 146, 170, 194, 230, 266, 302,
350, 434, 590, 770, 974, 1202, 1454, 1730, 2030, 2354, 2702,
3074, 3470, 3890, 4334, 4802, 5294.
If you want to use Gauss-Legendre angular points (a=2N^2)
instead of Lebedev numbers use BIGGRID=-rrraaaaaa
(i.e. use a negative number).
Return to Top
"6-31G* Basis Set for Third-Row Atoms", V. Rassolov,
J. A. Pople, M. Ratner, P. C. Redfern, and L. A. Curtiss,
J. Comp Chem., 22 (9) 976-984
"Gaussian-3 (G3) theory for molecules containing first and second-row atoms"
J. Chem. Phys. Vol. 109, No. 18, (Nov. 8 1998) pp 7764-7776
"Gaussian-3 theory using reduced Moller-Plesset order"
J. Chem. Phys. Vol 110, No. 10, (Mar. 8 1999) pp 4703-4709
"A Brief Guide to Molecular Mechanics and Quantum Chemical Calculations"
W.J. Hehre, J. Yu, P.E. Klunzinger, L. Lou;
Wavefunction Inc.
"A Guide to Molecular Mechanics and Quantum Chemical
Calculations"
W.J. Hehre;
Wavefunction Inc. (2003)
As of June 2008 this books ships with all versions of
Spartan as a
pdf file.
"Ab Initio Molecule Orbital Theory", Warren J. Hehre, Leo Radom,
Paul v.R. Schleyer, John A. Pople; John Wiley & Sons,
Inc. (1986)
"Efficient Calculation of Heats of Formation"
W.S. Ohlinger, P.E. Klunzinger, B.J. Deppmeier, W.J. Hehre,
J. Phys. Chem. A 2009 113 (10), 2165-2175
"1998 CODATA Recommended Values of the Fundamental Physical
Constants"
which derives the "..values of the basic constants and conversion factors
of physics and chemistry resulting from the 1998 least-squares adjustment
of the fundamental physical constants as ... published by the CODATA Task
Group on Fundamental Constants and as recommended for international use by
CODATA. The new, 1998 CODATA set of recommended values replaces its
predecessor published by the Task Group and recommended for international
use by CODATA in 1986..." and prior to that in 1973.
Yan Zhao, Nathan E. Schultz, and Donald G. Truhlar
(2006). "Design of Density Functionals by Combining the Method
of Constraint Satisfaction with Parameterization for
Thermochemistry, Thermochemical Kinetics, and Noncovalent
Interactions". The Journal of Chemical Theory and Computation
(ACS Publications) 2 (2):
364-382.
doi:
10.1021/ct0502763.
Yan Zhao and Donald G. Truhlar (2008). "The M06 suite of
density functionals for main group thermochemistry,
thermochemical kinetics, noncovalent interactions, excited
states, and transition elements: two new functionals and
systematic testing of four M06-class functionals and 12 other
functionals". Theoretical Chemistry Accounts (Springer Berlin /
Heidelberg) 120 (1-3): 215-241.
doi:
10.1007/s00214-007-0310-x.
Yan Zhao and Donald G. Truhlar (2008). "A new local density
functional for main-group thermochemistry, transition metal
bonding, thermochemical kinetics, and noncovalent
interactions". The Journal of Chemical Physics (American
Institute of Physics) 125 (8):
194101-194119.
doi:10.1063/1.2370993.
doi:
10.1063/1.2370993.
Yan Zhao and Donald G. Truhlar (2008). "Density Functional for
Spectroscopy: No Long-Range Self-Interaction Error, Good
Performance for Rydberg and Charge-Transfer States, and Better
Performance on Average than B3LYP for Ground States". The
Journal of Physical Chemistry A (ACS Publications) 110 (49):
13126–13130.
doi:
10.1021/jp066479k.
Jeng-Da Chai and Martin Head-Gordon (2006). "Systematic
optimization of long-range corrected hybrid density
functionals". The Journal of Chemical Physics (American
Institute of Physics) 128 (8):084106-084121.
doi:10.1063/1.2834918.
doi:
10.1063/1.2834918.
J. D. Chai and Martin Head-Gordon (2008). "Long-range corrected
hybrid density functionals with damped atom-atom dispersion
corrections". Physical Chemistry Chemical Physics (RSC
Publishing) 10 (44): 6615-66120.
doi:
10.1039/b810189b.
S.-H. Chien and P. M. W. Gill,
"SG-0: A Small Standard Grid for DFT Quadrature on Large Systems"
J. Comput. Chem., 27, 730, (2006)
P. M. W. Gill, B. G. Johnson and J. A. Pople,
"A standard grid for density functional calculations"
Chem. Phys. Lett. , 209, 506, (1993)
doi:
10.1016/0009-2614(93)80125-9.
Return to Top
Wavefunction Support
Author: Phil Klunzinger