The energy reported in mechanics is a "strain energy". The functional form can be found in J. Comp. Chem., (17) 1996, 490-641, but can be summarized as the strain in each bond, angle, and torsion, as well as the energy of non-bonded terms. It is important to note that this energy is relative to an unstrained version of the molecule being examined. Thus this energy cannot be compared with energy of other molecules. Even comparing energies of structural molecules is not appropriate with molecular mechanics. What molecular mechanics is good at is getting structures and conformational energy. More discussion on the appropriate use of Energies can be found in the A Guide to Molecular Mechanics and Quantum Chemical Calculations. A quick summary of this guide can be found in Selecting a Theoretical model & Calculation Times, as well as the Quantum Mechanics Energy Questions FAQ document.
Return to Top
At times the MMFF atom-typing algorithms cannot correctly type an atom. For example, an Iron ion with no bonds can be either Fe+2 or Fe+3. The FFHINT keyword is a way to bypass the atom-typing. The general format is:
| 'name' | is the label of the atom to change,
for example 'Fe1' or 'Fe2'. Characters are case insensitive, and if the last character of name is an asterisk (*) then name is treated as a wildcard. i.e. 'Fe*' matches 'Fe1', 'FE3', 'fe', and 'fe123'. |
| '~~' | are two tildes in a row |
| 'hint' | can be:
|
You can have up to 10 FFHINTs by adding a digit prior to the "equal sign", ie:
Use PRINT_FF for more information on how specific hints are used internally. Note that these are "hints" and may be ignored.
Return to Top
In Spartan, ligand points are used in the graphical user interface to represent multi-center ligand bonds. For example the "eta-5" of the cyclopentadiene anion metal bond found in metallocenes. It is important to note that while the "Ligand Point" is important in mechanics, it is not used in any of the ab initio methods.
- Ligand points:
- The default ligand pt. is type -1 (for use in params.MERCK)
and is defined as the center of ligand atoms
- If the ligand point is not used in any constraint it is, by definition, the center of its defining points.
- If the ligand point is used in a torsion or length constraint the ligand is constrained to the center using a quadratic force.
- eta bonds connecting the defining ligand atoms (ie: Ti--C in TiCp2) have not stretch-bend component.
- torsion angle force constant cannot use a ligand point
- Angle terms of the form a-M-Lig or Lig-M-Lig may be used
- eta bonds have a bond type (see params.MERCK) of '-n' where is the order of the bond. A default/fallback of -1 is used in bond lookup.
- no bond angle or bond torsion force constants use the eta bond.
- Note: Ligand points are an extension to the Merck Force-field.
- The default ligand pt. is type -1 (for use in params.MERCK)
and is defined as the center of ligand atoms
- Central atoms of high coordinate metals can be a D0 type.
- no torsion contain type 50 in internal (j,k),
- no a-D0-b bond angle, instead 1-4 VdW interactions are applied to a-b.
- Note: Metals are an extension to the MMFF94 Force field.
- summary example: Cp-M-Cl
- The Cp-M-Cl bond angle can be set (default for type 50: not set).
- The Cp-M distance is controlled by Carbon-M,eta-5 bonds. A Cp-M bond can be set in the param file
- The ligand is placed in the center of the Cp by a hookian spring.
- All other bond angles which include the ligand point or the new Carbon-M eta bonds are turned off.
- All torsions contain the Carbon-M bond are turned off.
- All torsions (i,j,k,l) with the ligand point at j or k are turned off.
- The special case the torsion of eta-2 bonds is not yet implemented.
Return to Top
The minimization method in mechanics is a "Truncated Newton Method" using a preconditioned conjugated Gradient (PCG) algorithm with an exact, sparse, hessian. In the general spirit of TNPACK Tamar Schlick & Aaron Fogelson in "ACM Transactions on Mathematical Software, Vol. 18, No. 1 March 1992."
Return to Top
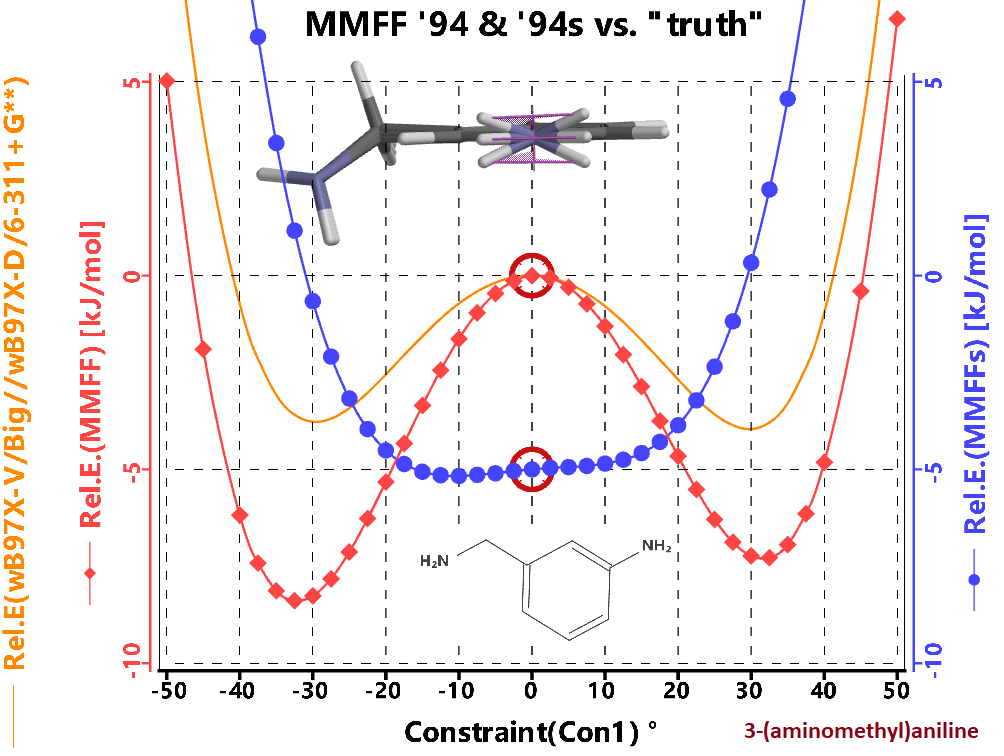 The only difference between MMFF94 and MMFF94s is that
the later flattens some Nitrogen groups attached to benzene rings.
The classical example is the NH2 group in aniline.
The only difference between MMFF94 and MMFF94s is that
the later flattens some Nitrogen groups attached to benzene rings.
The classical example is the NH2 group in aniline.
High-end calculations show that this NH2 group prefers to be slightly out-of-plane, however most experiments show that the time average location of the hydrogens is in the plane as the barrier to inversion is so low. The MMFF94s parameterization encourages this group to be planar. We recommend using MMFF94, especially since mechanic's primary use in Spartan is a starting point for higher end quantum mechanical calculations.
In the image at right we show the energy profile comparing a "good" DFT method with MFF94 and MFF94s for 3-(aminomethy)aniline. The 3 configurations shown are the two minima, and central maxima for the DFT method with the out-of-plane angle (C-H-H-N) being +-30, and 0 respectively. While MMFF94 overestimates the strength of the inversion barrier, the geometry closely matches the DFT method.
Return to Top